The voltage-gated sodium channel
|
The rapid depolarization (fase 0) of atrial and ventricular muscle cells and the His-Purkinje system depends on the activation of the fast inward Na+ current (INa) through voltage-gated Na+ channels. INa underlies the initiation and propagation of AP and determines cardiac excitability and conduction velocity of electrical stimuli through the heart (George 2005). Voltage-gated Na+ channels are also important targets for local anesthetics and class I antiarrhythmic agents.
Voltage-gated cardiac Na+ channels contain an α subunit (Nav1.5, 260 kDa) encoded by SCN5A gene and one or more α-subunits encoded by SCN1B-SCN4B genes. Most cardiac Na+ channels are tetrodotoxin (TTX)-resistant (KD = 2–6 μM) channels. Other α subunits typical of neuronal Na+ channels (Nav1.1, Nav1.3 and Nav1.6) sensitive to tetrodotoxin (KD = 1-10 nM) are also expressed in the myocardium which may play a role in sinus node pacemaker activity (Lei et al., 2007). The subcellular localization of Nav1.5-encoded myocardial Nav channels at the intercalated disks suggests that these channels play a major role in regulating conduction (Kucera and Rudy, 2002).
The pore-forming α subunit is sufficient for functional expression, but the kinetics and voltage-dependence of channel gating are modified by the β subunits and these auxiliary subunits are involved in channel localization and interaction with cell adhesion molecules, extracellular matrix and intracellular cytoskeleton (Caterall et al., 2019). Four different β subunits (33-36 kDa) encoded by SCN1b, SCN2b, SCN3b and SCN4b genes, respectively, are type 1 transmembrane glycoproteins with a single transmembrane domain, an extracellular N-terminus and a cytoplasmic C-terminal (Brackenbury e Isom, 2011). The extracellular end has a structure homologous to the immunoglobulin superfamily (IgG), suggesting that subunits may act as cell adhesion molecules that target channels to the plasma membrane and mediate channel interactions with a variety of signaling molecules (Makita et al., 1996; Isom, 2001; Brackenbury and Isom, 2011). Moreover, β subunits interact with various extracellular matrix proteins from the cytoskeleton (ankyrins that regulate the trafficking of a variety of plasma membrane proteins), transmembrane proteins (cadherins, connexins) or involved in intracellular trafficking from the endoplasmic reticulum to the sarcolemma (e.g., glycerol-3-phosphate dehydrogenase), or with enzymes (Ca2+/calmodulin-dependent protein kinase II or CaMKII) (Xiao et al., 1999; Ratcliffe et al., 2001; Mohler et al., 2002). Furthermore, the interaction between α and β1.1 subunits decreases the affinity of local anesthetics and antiarrhythmic drugs for cardiac Na+ channels (Makielski et al., 1996). Finally, mutations in genes encoding β-subunits are present in various primary arrhythmogenic syndromes, confirming their important role in the regulation of cardiac Na+ channels. Many proteins interact with Na+ channels transiently in the context of channel regulation, including several protein kinases and G proteins, while othersinteract more permanently, including cell adhesion molecules and cytoskeletal proteins (Caterall et al., 2019). The ubiquitous Ca2+-dependent regulator calmodulin binds to a conserved site in the proximal C-terminal domain of many Na+ channels and serves as a target for calcium regulation (Gabelli et al., 2014). Fibroblast Growth Factor Homologous Factors (FHFs) can also form a tight complex with the proximal C-terminus of Na+ channels and mutations localized in the central binding core on Nav1.5 for FHFs (p.H1849R) produce a gain-of-function for INa by altering steady-state inactivation and slowing the rate of Nav1.5 inactivation and myocytes expressing Nav1.5 p.H1849R displayed prolonged action potential duration and arrhythmogenic afterdepolarizations (Musa et al., 2015).
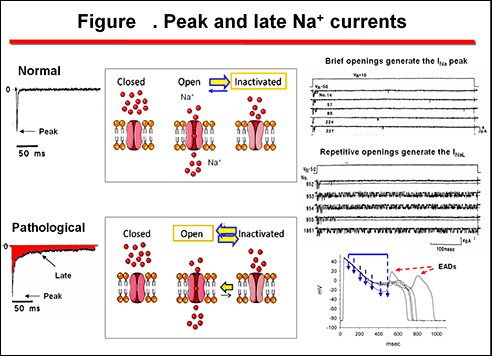
During the action potential, Na+ channels switch between three conformational states (Caterall, 2000; Balser, 2001)(figure 2). The voltage- and time-dependent transition between these conformational states is referred to as gating. During the diastole the channel is in the resting (R) nonconducting conformation. During cardiac depolarization (phase 0 of the AP) there is a rapid rise in Na+ permeability due to the opening (activation) of the channels from their resting state (R → O), allowing Na+ entry (INa). Models of voltage sensor function predict that the S4 segment slides 6-8 Å outward through a narrow groove formed by the S1, S2, and S3 segments, rotates ∼ 30°, and tilts sideways at a pivot point formed by a highly conserved hydrophobic region near the middle of the voltage sensor. The S4 segment has a 3(10)-helical conformation in the narrow inner gating pore, which allows linear movement of the gating charges across the inner one-half of the membrane. Conformational changes of the intracellular one-half of S4 during activation are rigidly coupled to lateral movement of the S4-S5 linker, which could induce movement of the S5 and S6 segments and open the intracellular gate of the pore (Yarov-Yarovoy et l., 2012).
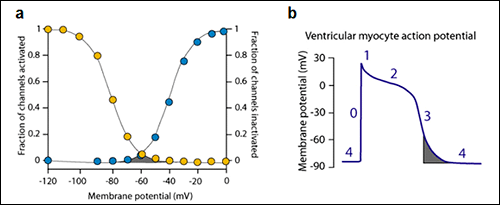
Additionally, a very small fraction of Na+ channels may reactivate during the phase 3 of the action potential. The probability of Na+ channel opening during the phase 3 of the cardiac AP is determined by the crossover of the activation and inactivation curves which govern the opening of the sodium channel (Figure xa) (Attwell et al., 1979). At the membrane potentials at which this crossover occurs a fraction of channels have recovered from inactivation and may reopen generating a new propagated response at the end of phase 3 of repolarization (Figure xb). The contribution of this window current under physiological circumstances is very limited because the overlap is less than 5% of the maximum current in healthy myocytes. However, under pathological conditions, some mutations of the Na+ channels can shift of voltage dependence of steady-state inactivation toward more depolarized membrane potentials leading to an increased “window” inward sodium current that can can lead to early afterdepolarizations arising at the end of the phase 3 of the cardiac AP (Amin et al., 2010).
Figure . The window sodium current. a) The window current (gray area) is determined by the crossover of the activation and inactivation curves which govern the opening of the sodium channel. B) This crossover takes place during the late phase 3 of repolzarization.
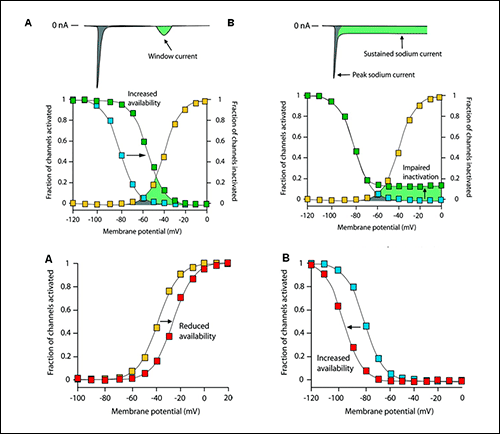
Figure. Effects of sodium channel mutations on channel gating. |
|
|
| Aviso legal Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional |
