Síndrome de QT Largo Congénito (SQTLC)
|
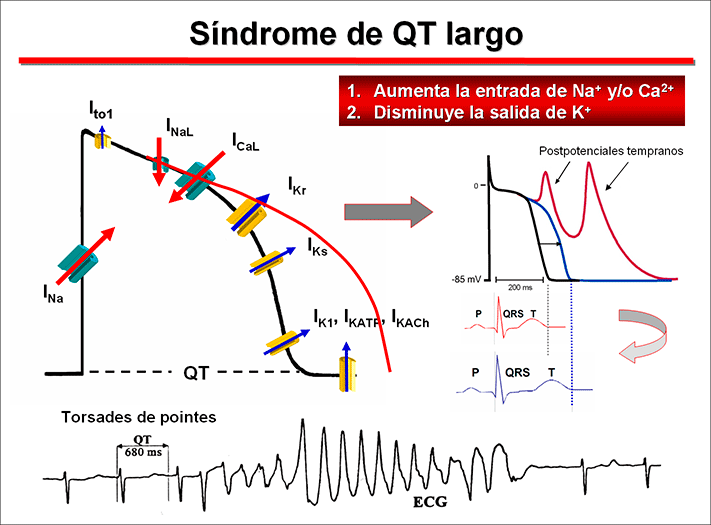
Síndrome de QT largo (SQTL) comprende un grupo de trastornos de la repolarización cardiaca caracterizados por una prolongación excesiva y heterogénea del PA ventricular y del intervalo QT corrregido por la frecuencia cardiaca en el ECG de la superficie (QTc Bazzet >440 ms en hombres y >460 ms en mujeres) que predispone a los sujetos afectados a la aparición de síncope, convulsiones y TV polimórficas (torsades de pointes) y aumenta el riesgo de SCD (Goldenberg y Moss, 2008). El síncope y las convulsiones asociadas con el SQTL son causados por torsade de pointes, una TV polimórfica caracterizada por la torsión del eje del QRS alrededor de la línea isoeléctrica de un trazado de ECG. Aunque la torsade de pointes son generalmente autolimitadas, a veces pueden degenerar en FV y SCD. ¿Quiénes tienen un mayor riesgo de padecer SQTLc? Por lo general afecta a niños y adultos jóvenes aparentemente sanos y la edad media de inicio de síntomas (síncope o SCD) son los 12 años de edad o incluso antes en las formas más graves de SQTL (Goldberg y Moss, 2008; Priori et al, 2003). Existen dos variantes fenotípicas del SQTLc. En 1957, Jervell y Lange Nielsen describieron el síndrome que lleva su nombre, que cursa neurosensorial congénita, intervalo QT largo en el ECG, mala respuesta a β-bloqueantes y una elevada incidencia de muerte súbita por arritmias ventriculares en los primeros años de vida. El patrón de herencia es autosómico recesivo. El síndrome de Jervell-Lange Nielsen aparece en 1.6-6 por millón de nacidos (Schwartz et al., 2006). Entre 1963 y 1964, Romano y Ward describieron de forma independiente una alteración cardiaca similar (QT largo y MSC) que no cursaba con sordera congénita. Este síndrome 1 de cada 2.000-5.000 nacidos vivos, con un ligero predominio de mujeres, y parece heredarse de forma autosómica dominante, pero con una penetrancia muy variable (25-90%). La tasa de mortalidad es del 21% para los pacientes sintomáticos que no reciben terapia dentro de un año a partir del primer síncope (Schwartz et al., 2012). Sin embargo, dado que el 10-35% de los pacientes tienen un intervalo QT (QTc) corregido normal (36% de los pacientes con LQT1, 19% con LQT2, y 10% with LQT3), es posible que la prevalencia real sea mucho mayor. De hecho, hasta un 30% de los portadores de la mutación pueden cursar con un QTc normal, pero son más susceptibles a desarrollar arritmias malignas que el resto de la población. La principal característica del SQTL es una marcada prolongación del intervalo QT del ECG asociada a una prolongación no uniforme en la duración de los PA ventriculares y del intervalo QT corregido (QTc) del ECG que resulta de un aumento en las corrientes despolarizantes de entrada de de Na+ y Ca2+ y/o una disminución en las corrientes repolarizantes de salida de K+. Esta prolongación de la DPA es más marcada en el epicardio que en el endocardio, lo que facilita la dispersión transmural de la repolarización (DTR) y de la recuperación de la excitabilidad ventricular y la aparición de pospotenciales tempranos que aparecen durante la fase 2 (meseta) y el comienzo de la fase 3 del PA ventricular. Estos pospotenciales tempranos que aparecen durante las fases 2 y 3 del potencial de acción pueden deberse a: 1) que la prolongación de la fase de meseta permite la reactivación-reapertura de los canales de Ca2+ tipo-L, 2) tiene lugar una reapertura repetitiva de los canales de Na+ durante la fase de meseta, lo que genera la corriente lenta de entrada de Na+ (INaL) y 3) la activación de activación de la corriente generada por el intercambiador Na+-Ca2+ (INCX). El inicio de pospotenciales tempranos en presencia de un aumento en la dispersión de la refractariedad entre las diversas regiones ventriculares modifica el patrón de conducción ventricular, pudiendo producir áreas de bloqueo unidireccional y conducción lenta, condiciones que pueden facilitar la reentrada ventricular. Por otra parte, la aparición de alternacia en la DPA, atribuida a una mayor pendiente de la curva de restitución de la APD junto con el gradiente de repolarización anormal también puede conducir a un bloqueo unidireccional de la conducción y, por lo tanto, a la reentrada. Sin embargo, en el SQTL, la velocidad de conducción o el periodo refractario ventriculares apenas si se modifican.
1. Síndrome de Romano-Ward. El síndrome de Romano-Ward se caracteriza por una gran heterogeneidad genética. Se han identificado, al menos, 16 loci distintos que si bien cursan con el mismo fenotipo (prolongación del QTc y aumento en la dispersión de la repolarización ventricular que facilitan la aparición de pospotenciales tempranos y de arritmias por reentrada), presentan una fisiopatología, curso clínico, pronóstico y tratamiento distintos.
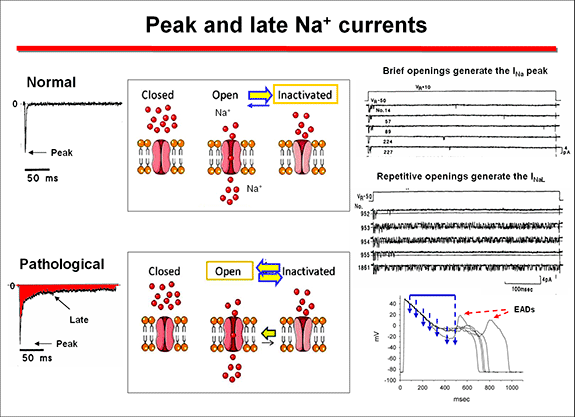
El SQTL1 se asocia con mutaciones en el gen KCNQ1. Las mutaciones localiazadas en los segmentos transmembranas están relacionadas con un QTc más largo, eventos cardíacos más frecuentes y una mayor prolongación del QTc con el ejercicio (Jons et al., 2009). Las mutaciones en los lazos intracitoplasmáticos afectan a los sitios de fosforilación mediada al aumentar el tono simpático y se asocian específicamente con una prolongación del QT durante el ejercicio, aunque el QT puede ser normal en reposo. Las mutaciones en los bucles citoplásmicos de la proteína KCNQ1 o las mutaciones que producen un efecto dominante negativo se asocian con un peor pronóstico, especialmente cuando se comparan con las mutaciones que afectan a las regiones C-terminales de la proteína, pero responden más favorablemente a los b-bloqueantes (Goldenberg et al., 2006; Jons et al., 2009). El SQTL2 se asocia con mutaciones con pérdida de función en el gen KCNH2 y da como resultado una reducción de la IKr, un retraso de la repolarización y una prolongación del intervalo QT. Las mutaciones homocigotas dan como resultado la muerte intrauterina y si el individuo sobrevive presenta una marcada prolongación del QT. Por lo tanto, las mutaciones heterocigotas en HERG son la norma en pacientes con SQTL2. Las mutaciones con cambio de sentido asociadas al SQTL2 pueden causar la pérdida de la función por varios mecanismos (Anderson et al, 2008; Sanguinetti, 2010): 1) por producir un plegamiento defectuoso de la proteína. Las proteínas mal plegadas se retienen en el retículo endoplásmico (ER) y las subunidades allí retenidas se degradan rápidamente por la vía ubiquitina-proteasoma. Este es el caso de las mutaciones HERG Y611H y V822M (Zhou et al., 1998). 2) Interrumpir el tráfico desde el RE hacia el aparato de Golgi y la membrana superficial. 3) Alterar las características de la activación de canales hERG. Las mutaciones que aceleran la inactivación (p. ej., G584S) (Zhao et al., 2009) o la velocidad de desactivación (p. ej., M124R u otras mutaciones en el dominio PAS) (Sushi et al., 2005) reducen la IKr durante la repolarización de la PA. 4) Otras mutaciones (I593R y G628S) se procesan de manera similar a la proteína hERG nativa, pero no producen canales funcionales. Otras mutaciones sin sentido causantes del LQT2 (W1001X, R1014X) disminuyen los niveles de ARNm mutado al inducir su degradación mediada por mutaciones terminadoras, un mecanismo celular de vigilancia del ARNm para detectar mutaciones terminadoras (las que crean nuevos codones de paro) y evitar la expresión de proteínas truncadas o erróneas (Gong et al., 2007). Con la excepción de la asociación infrecuente con la epilepsia, las mutaciones de KCNH2 no producen enfermedades en otros órganos además del corazón. Las mutaciones que ocurren en los canales de K+ pueden causar una pérdida de función y una disminución de las corrientes de salida de K+ porque: 1) las subunidades de proteínas mutantes y de tipo salvaje pueden unirse y ejercer un efecto negativo dominante sobre la corriente; 2) algunas subunidades mutantes pueden no unirse con los péptidos de tipo nativo, lo que resulta en una pérdida de función que se reduce en ≤50% (haploinsuficiencia); 3) las mutaciones interfieren con el tráfico de subunidades intracelulares, impidiendo que los péptidos mutados alcancen la membrana celular; 4) cambios en la apertura-cierre (compuerta) del canal, y 5) cambios en la respuesta del canal a la estimulación simpática y/o en la nitrosilación del canal iónico (Sanguinetti, 2010). SQTL3. Comprende entre el 5% y el 10% de los pacientes con SQT (la incidencia se estima en 1: 20,000). El SQTL3 resulta de mutaciones con ganancia de función (la mayoría son mutaciones sin sentido) en el gen SCN5A por al menos 4 mecanismos distintos (ver Clancy y Kass, 2005).
2) Con menos frecuencia las mutaciones facilitan la reapertura del canal previemente inactivado generando una corriente de Na+ ("window current") en el rango de potenciales a los que la activación y la inactivación del canal se superponen que puede generar generar pospotenciales tempranos al final de la fase de repolarización (Amin et al., 2010). La INa window aumenta cuando la inactivación de los canales mutados se produce a niveles más despolarizadas,
mientras que la activación no se modifica, lo que aumenta el rango de voltajes a los que el canal puede reactivarse y generar la INa (Wang et al., 1996). 2.Síndrome de Jervell y Lange Nielsen. Está causado por mutaciones homocigotas o heterocigotas compuestas en los genes KCNQ1 y KCNE1 que codifican las subuniddaes que generan la IKs(Schwartz et al., 2006). Las mutaciones no muestran efectos dominantes negativos, lo más probable porque las proteínas mutantes no son estables o no formen heteromultímeros con subunidades nativas. La mayoría de las mutaciones (80%) están en el gen KCNQ1. Los β-bloqueantes presentan una eficacia parcial, por lo que el 51% de los pacientes tuvieron eventos a pesar del tratamiento y el 27% presenta paro cardíaco y MSC. Los subgrupos con menor riesgo son las mujeres, los pacientes con QTc ≤ 550 ms y aquellos sin eventos en el primer año de vida o con mutaciones en KCNE1 (Schwartz et al., 2006). El SQTL presenta una penetrancia baja y muy variable (55%, 70% y 80% en los SQT1, SQT2 y SQT3, respectivamente), lo que indica que 2 de cada 5 portadores de una mutación tienen un QTc normal (Priori et al., 1999, 2003). Esto ocurre incluso en pacientes con historia previa de síncope, aunque ellos son más susceptibles a desarrollar arritmias ventriculares malignas que el resto de la población. Algunos pacientes pueden permanecer asintomáticos durante mucho tiempo, de modo que la MSC puede ser la primera manifestación clínica de un individuo joven "aparentemente sano". Otros pacientes presentan mareos o sensación de desvanecimiento (presíncope), síncope y convulsiones y la MSC. El síncope puede comenzar a edades tempranas de la vida, a menudo en la infancia (antes de los 10 años, lo que indica un curso maligno), pero la frecuencia y la intensidad de los síntomas pueden disminuir al llegar a la edad adulta. La aparición de un síncope durante la realización deuna actividad física o emoción fuerte es un signo que sugiere que el paciente puede presentar un SQTL; cuando además hay un historial familiar de síncope inexplicable o de SCD, la posibilidad de que nos enfrentemos a un SQTL aumenta significativamente. Cuando las convulsiones se producen en niños, éstas pueden ser diagnosticados erróneamente de crisis epilépticas. Por lo tanto, se recomienda realizar un ECG en todos los niños y jóvenes que han tenido un episodio de síncope y/o convulsiones de causa desconocida. Desafortunadamente, muchas veces sí se realiza un electroencefalograma (EEG), pero raramente un ECG. Factores que desencadenan los eventos cardiacos. Son específicos del gen y de la mutación. ¿Cómo se diagnostica el SQTLc? El hallazgo de que existe una relación entre edad, sexo, duración del QTc, historia familiar, genotipo y curso clínico, permite estratificar el riesgo del paciente y seleccionar el tratamiento médico más adecuado. A continuación describimos los criterios diagnósticos:
Se considera que los pacientes con ≥ 3.5 puntos tiene una alta probabilidad de presentar un evento. Sin embargo, asi un 50% de los pcientes con SQTL no presentan una marcada prolongación del intervalo QT del ECG (Sy et al., 2011) y portadores de mutaciones silentes (que presentan un QTc normal en reposo) representan el 36%, 19% y 10% de los pacientes con SQTL1, SQTL2 y SQTL3, respectivamente (Priori et al., 2003). Características electrocardiográficas
El ECG es la mejor forma de diagnosticar el SQTLc, ya que en muchos pacientes nos mostrará un intervalo QT prolongado. No obstante, el ECG puede ser normal, incluso en pacientes con historia de síncopes. Se considera como prolongado un intervalo superior a 440 milisegundos, fronterizo o límite cuando existe una prolongacion del QT de entre 450 a 470 milisegundos, y patológico cuando el QT es superior a 480 milisegundos en mujeres o 470 en hombres. La duración del QT puede variar en cada paciente, incluso llegando a ser normal en algunas circunstancias (aunque siguen presentado riesgo de síncopes por arritmias ventriculares). La presencia de QTc >500 mseg (en ausencia de síncope o antecedentes familiares) en más de una ocasión es suficiente para el diagnóstico de LQTS. Siin embargo, la precisión en el diagnóstico del SQTL se ve dificultada por la observación de que los pacientes pueden tener valores de QTc dentro de los límites normales, incluso si son genotipo positivos, o pueden mostrar una prolongación de QT solo en circunstancias específicas, lo que da lugar a una posible superposición significativa en el QTc entre los controles sanos y los pacientes con LQTS. . Un QTc> 445 ms a 4 minutos en recuperación después del ejercicio aumenta la probabilidad de LQTS con mayor especificidad.
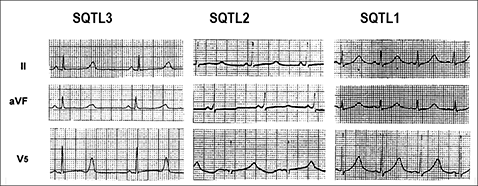
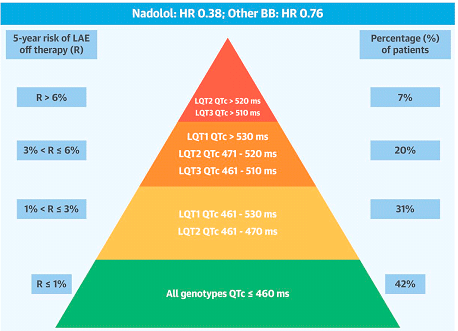
Otras características ECG son (Moss et al., 2002, 2007, Schwartz et al., 1993, 2006, Zhang et al., 2000): 1) una mayor dispersión del intervalo QT, que refleja las diferencias regionales en la repolarización ventricular. La repolarización en el corazón es normalmente heterogénea debido a la diferencia en la duración del potencial de acción en las diferentes regiones (ventrículo derecho vs. izquierdo o epicardio us. endocardio) como consecuencia de la distinta expresión de los diversos canales iónicos. La dispersión del intervalo QT es de 48 ± 18 mseg en sujetos normales y este valor aumenta de forma significativa en pacientes con SQTLc, estando este incremento relacionado con un riesgo mayor de presentar arritmias ventriculares malignas. 2) Alteraciones de la onda T: con alternancia en la polaridad, apariencia bifásica, variaciones en la amplitud o muescas. La alternancia de la onda T (variación latido a latido de la amplitud, morfología y polaridad de la onda T en ritmo sinusal, sin variaciones en el complejo QRS) es un indicador de inestabilidad eléctrica, que refleja la dispersión regional en la repolarización y, en ocasiones, precede a la FV. 3) Signos de disfunción del nodo sinusal (SQTL4), bradicardia sinusal (SQTL1 y SQTL3) y/o pausas sinusales. 4) Bloqueo AV 2:1 aparece en el 4-5% de los pacientes, particularmente en los que presentan SQTL2, SQTL3 y SQTL8, y se asocia a una alta mortalidad a pesar del tratamiento con β-bloqueantes y/o un DAI. 5) Muchos episodios de torsades de pointes están precedidos por disminuciones repentinas en la frecuencia cardiaca o de una pausa seguida de una extrasístole: intervalo RR "corto–largo–corto" (Viskin et al., 2000). Una pausa eléctrica aumenta el intervalo QT y la heterogeneidad de la repolarización ventricular y la aparición de un latido prematuro durante la parte vulnerable de la repolarización pueden precipitar la TV. A pesar de la heterogeneidad genética, 3 genes explican >90% de los casos de SQTL: KCNQ1 (SQTL), KCNH2 (SQTL) y SCN5A (SQTL). La evolución de los pacientes con SQTL es muy variable, dependiendo de la edad, el sexo, la duración del intervalo QTc, el genotipo del SQTL y la respuesta al tratamiento (Priori et al., 2003). Se considera que el riesgo es bajo (<30%) en pacientes con un QTc <500 ms, si son varones con un SQTL2 o en pacientes con una mutación causante de un SQTL1 o SQTL2 (Figura). El riesgo es intermedio (30-50%) en pacientes con un QT <500 ms si son varones con un SQTL3 o mujeres con un SQTL2 o un SQTL3 o si son mujeres que presentan un QTc 500 m y un SQTL3. El riesgo es alto (>50%) incluye a mujeres con un SQTL2 y varones con un SQTL3 que presentan un QTc ≥ 500 ms independientemente de otros factores de riesgo (Priori et al., 2003). La frecuencia de eventos clínicos antes del inicio de la terapia con β-bloqueantes desde el nacimiento hasta los 40 años de edad es significativamente mayor en pacientes con un SQTL2 (46%) y SQTL3 (42%) en comparación con aquellos con un SQTL1 (30%). También hay evidencia de que los eventos en pacientes con un SQTL3 tienden mayor probabilidad de ser letales y parecen ocurrir más tarde en la infancia, durante o después de la pubertad. Figura. Riesgo de 5 años de eventos arrítmicos que ponen en peligro la vida del paciente (LAE) según el genotipo y intervalo QTc antes del tratamiento. En la parte superior se muestra el efecto de los b-bloqueantes (Mazzanti et al., 2018). La columna de la izquierda indica el riesgo a 5 años de ELA que corresponde a cada grupo de pacientes con códigos de colores caracterizados por la duración del QTc y el genotipo (de verde: menor riesgo; rojo: mayor riesgo). La columna de la derecha indica el porcentaje de pacientes en cada categoría codificada por colores presentes en la cohorte. BB: b-bloqueantes. HR: relación de riesgo. LAE: eventos arrítmicos que amenazan la vida; .QTc: intervalo QT corregido.
Las pruebas genéticas tienen implicaciones diagnósticas, pronósticas y terapéuticas en pacientes con SQTL. Las mutaciones con cambio de sentido (missense mutations) son las más comunes (78%, 67%, y 89% en KCNQ1, KCNH2 y SCN5A) y su localización determina el riesgo del paciente; la mutaciones sin sentido tienen un valor predictivo >99% independientemente de su localización (Kapa et al., 2009).
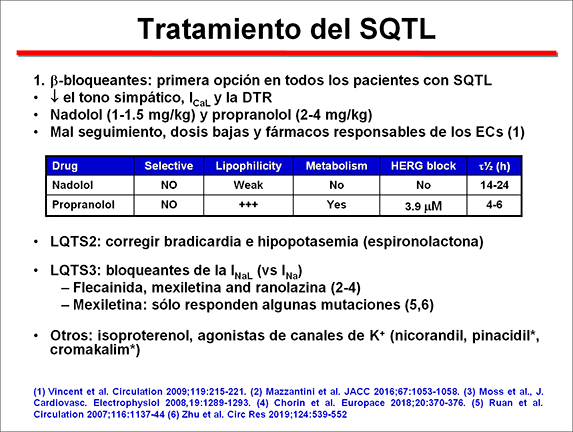
Hay cuatro niveles de tratamiento del LQTS, aplicados de acuerdo con la gravedad del cuadro, pero los bloqueantes -adrenérgicos y el desfibrilador cardioversor implantable (ICD) representan la primera opción de terapia LQTS (Priori et al., 2015).
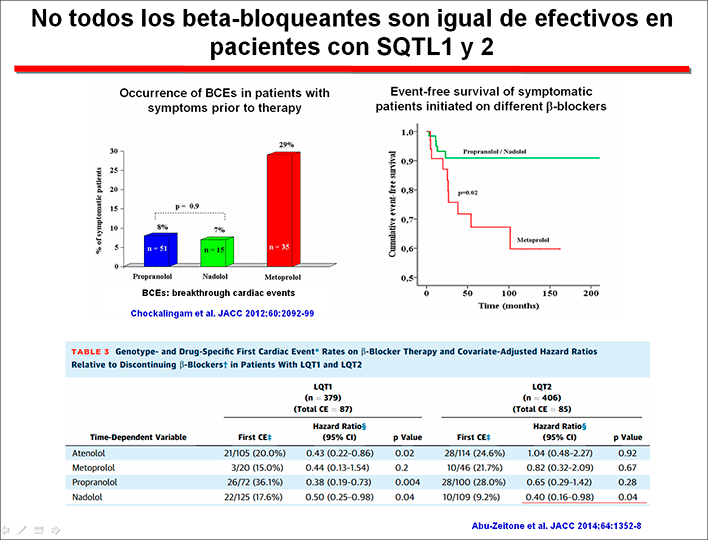
El efecto protector de los β-bloqueantes es desconocido, pero se ha correlacionado con la supresión del tono simpático que es uno de los factores desencadenantes de las torsades de pointes, el bloqueo de los canales de calcio de tipo-L y la disminución de la dispersión transmural del intervalo QTc (Moss et al, 2000; Gemma et al, 2011). Sin embargo, no todos los β-bloqueantes son igualmente eficaces. El propranolol y el nadolol son más efectivos que el atenolol y el metoprolol y el cambio de propranolol o nadolol al metoprolol se ha asociado con recurrencias trágicas (Chockalingam et al., 2012; Schwartz et al, 2012). Los datos con atenolol son limitados.
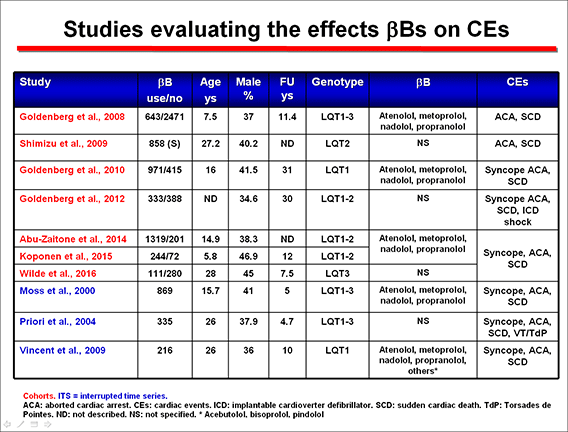
Los resultados de estos estudios fueron analizados por Ahn et al (2017). Podemos ver que los β-bloqueante: 1) reducen de forma asignificativa los eventos cariacos (síncope, parada cardiaca abortada y MSC). 2) Su efecto aparecía en todos los genotipos, aunque como ya hemosmencionado el efecto en pacientes con SQTL3 era muy evidente en mujeres. 3) Existían diferencias entre los distintos β-bloqueantes. El nadolol era muy efectivos en pacientes con SQTL1 o 2, mientras que propranolol y atenolol eran sólo efectivois en el SQTL1, pero no en el SQTL2 y el metoprolol no era efectivo en ningún genotipo.
Otros tratamienos farmacológicos.
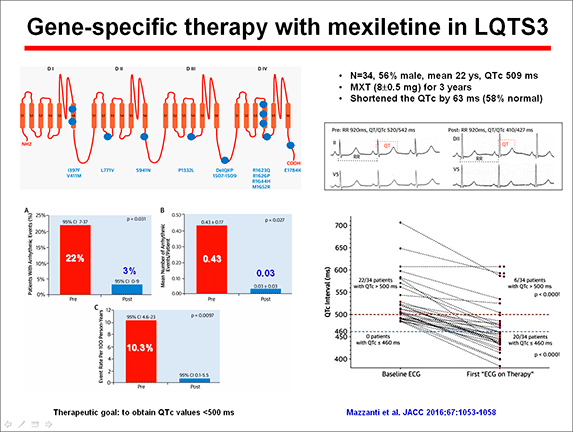
1) bloqueantes de los canales de Na+. En pacientes con SQTL3, mexiletina, flecainida y ranolazina acortan el intervalo QTc en pacientes con SQTL3 por sus propiedades bloqueantes de los canales de Na+ (Schwartz et al., 1995). Estos fármacs bloquean más la INaL que la INa pico. Sin embargo, el efecto puede ser específico de la mutación y la mayor parte de la experiencia clínica se ha obtenido con la mexiletina. Sin embargo, a altas concentraciones, estos fármacos también pueden bloquear el INa pico y ejercer efectos pro-arrítmicos al disminuir la excitabilidad cardíaca y disminuir la velocidad de conducción.
In vitro, el efecto de la mexiletina está influenciado por las propiedades biofísicas de la mutación específica, de modo que la mexiletina acorta el intervalo QTc en pacientes con mutaciones de SCN5A que desplazan la inactivación hacia potenciales más negativos (es decir, inactivación más temprana) (R1626P, P1332L y R1626P) pero no es efectiva en otras mutaciones (S941N, M1652R) (Ruan et al., 2007, 2010). La mexiletina se puede administrar en el hospital a pacientes con SQTL3 y si el fármaco acorta el QTc> 40 ms, se recomienda asociarla al tratamiento -bloqueantes (Schwartz et al., 2012; Chockalingam et al., 2012). Muy recientemente, Zhu et al (2019) analizaron la relación entre el bloqueo de mexiletina y las propiedades biofísicas del canal y encontraron que la mexiletina alteraba la conformación del dominio de detección de voltaje (VSD) del canal cardiaco Na+ en el dominio III, que es el mismo VSD que muchas mutaciones asociadas al SQTL3 afectan.
La ranolazina, un nuevo fármaco antianginoso que bloquea de forma selectiva la corriente lenta de entrada de sodio (INaL) en los canales mutados asociados al SQTL3 (ΔKPQ e Y1795C) (IC50 = 15 vs 135 μM). El efecto de la ranolazina se reduce significativamente cuando se produce una mutación en un residuo (F1760A) que contribuye de manera crítica al sitio de unión para los anestésicos locales (LA) en el canal de Na+, lo que confirma que la ranolazina interactúa con el sitio del receptor para LA en el canal de sodio. En pacientes con SQTL3, la ranolazina acorta la duración del intervalo QT a concentraciones a las que no modifica la frecuencia, la contractilidad o la velocidad de conducción cardiacas (Moss et al., 2008).
3. Denervación simpática cardíaca izquierda (LCSD: left cardiac sympathetic denervation). La LCSD es especialmente efectiva en pacientes de alto riesgo en los cuales los β-bloqueantes no son efectivos en la prevención del síncope/arritmias, no son tolerados (QTc > 500 ms y SQTL2 y 3), no se aceptan o están contraindicados (pacientes asmáticos) y/o en quienes tienen un DAI está contraindicado o se rechaza ”(Priori et al., 2015). Es útil para pacientes en los que se producen eventos recurrentes de síncope y arritmias ventriculares graves a pesar del uso de β-bloqueantes o tras la implantación de un DAI. La LCSD ejerce un efecto antiarrítmico porque interrumpe la liberación de noradrenalina de los terminales cardíacos simpáticos y aumenta el umbral de fibrilación ventricular y la refractariedad ventricular. En 147 pacientes con SQTL seguidos durante 8 años, la denervación simpática cardíaca izquierda reducía los eventos cardíacos en ~ 90% (Schwartz et al., 2004). Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175-87. Abu-Zeitone A, Peterson DR, Polonsky B, et al. Efficacy of different beta-blockers in the treatment of long QT syndrome. J Am Coll Cardiol 2014;64:1352–8. Ackerman MJ, Mohler PJ. Defining a new paradigm for human arrhythmia syndromes:phenotypic manifestations of gene mutations in ion channel- and transporter-associated proteins. Circ Res 2010;107:457–465. Ahn J, Kim HJ, Choi JI, et al. Effectiveness of beta-blockers depending on the genotype of congenital long-QT syndrome: A meta-analysis. PLoS One. 2017;12:e0185680. Amin A, Asghari A, Tan HL. Cardiac sodium channelopathies. Pflugers arch 2010;460:223-237. Anderson CL, Delisle BP, Anson BD, et al. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. Bankston JR, Kass RS. Molecular determinants of local anesthetic action of beta-blocking drugs: implications for therapeutic management of long QT syndrome variant 3. J Mol Cell Cardiol 2009; 48:246-253.
Bennett PB, Yazawa K, Makita N, et al. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685 Chockalingam P, Crotti L, Girardengo G, et al. Not All Beta-Blockers Are Equal in the Management of Long QT Syndrome Types 1 and 2.J Am Coll Cardiol. 2012;60:2092-2099. Clancy CE, Kass RS. Inherited and acquired vulnerability to ventricular arrhythmias: cardiac Na+ and K+ channels. Physiol Rev. 2005;85:33-47. Compton SJ, Lux RL, Ramsey MR, et al. Genetically defined therapy of inherited long-QT syndrome. Correction of abnormal repolarization by potassium. Circulation. 1996;94:1018-22. Crotti L, Johnson CN, Graf E, De Ferrari GM et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 2013;127:1009-1017. Curran ME, Splawski I, Timothy KW, et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. Deo R, Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation. 2012;125:620–37. Epstein AE, DiMarco JP, Ellenbogen KA, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices); American Association for Thoracic Surgery; Society of Thoracic Surgeons. ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices): developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. Circulation. 2008;117:e350–e408. Etheridge SP, Compton SJ, Tristani-Firouzi M, et al. A new oral therapy for long QT syndrome: long-term oral potassium improves repolarization in patients with HERG mutations. J Am Coll Cardiol. 2003;42:1777–1782. Gemma LW, Ward GM, Dettmer MM, Ball JL, Leo PJ, Doria DN, Kaufman ES. β-Blockers protect against dispersion of repolarization during exercise in congenital long-QT syndrome type 1. J Cardiovasc Electrophysiol. 2011;22:1141–1146. George Jr AL. Common genetic variants in sudden cardiac death. Heart Rhythm. 2009;6(11 Suppl):S3–9. Goldenberg I, Mathew J, Moss AJ, et al. Corrected QT variability in serial electrocardiograms in long QT syndrome: the importance of the maximum corrected QT for risk stratification. J Am Coll Cardiol. 2006;48:1047–1052. Goldenberg I, Moss AJ, Peterson DR, et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation 2008;117:2184–91. Goldenberg I, Bradley J, Moss A, et al. Beta- blocker efficacy in high-risk patients with the congenital long-QT syndrome types 1 and 2: implications for patient management. J Cardiovasc Electrophysiol 2010;21:893–901. Goldenberg I, Thottathil P, Lopes CM, et al. Trigger-specific ion-channel mechanisms, risk factors, and response to therapy in type 1 long QT syndrome. Heart rhythm: the official journal of the Heart Rhythm Society. 2012;9:49–56. Gong Q, Zhang L, Vincent GM, et al. Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long-QT syndrome. Circulation 2007;116:17–24. Jons C, Moss AJ, Lopes CM, et al. Mutations in conserved amino acids in the KCNQ1 channel and risk of cardiac events in type-1 long-QT syndrome. J Cardiovasc Electrophysiol. 2009;20:859-65. Kapa S, Tester DJ, Benjamin BS, et al. Distinguishing Pathogenic Mutations From Benign Variants. Genetic Testing for ong-QT Syndrome.Circulation 2009;120:1752-1760 Kawakami K, Nagatomo T, Abe H, et al. Comparison of HERG channel blocking effects of various beta-blockers – implication for clinical strategy. Br J Pharmacol. 2006;147:642–652. Koponen M, Marjamaa A, Hiippala A, et al. Follow-Up of 316 Molecularly Defined Pediatric Long-QT Syndrome Patients. Circulation: Arrhythmia and Electrophysiology. 2015;8:815–23 Makita N, Behr E, Shimizu W, et al. The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J Clin Invest. 2008;118:2219–2229. Mazzanti A, Maragna R, Faragli A, et al. Gene-Specific Therapy With Mexiletine Reduces Arrhythmic Events in Patients With Long QT Syndrome Type 3. J Am Coll Cardiol. 2016 Mar 8;67(9):1053-8. Mazzanti A, Maragna R, Vacanti G, et al. Interplay Between Genetic Substrate, QTc Duration, and Arrhythmia Risk in Patients With Long QT Syndrome. J Am Coll Cardiol 2018;71:1663-1671. Marx SO, Kurokawa J, Reiken S,et al. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 2002;295:496–499. Medeiros-Domingo A, Kaku T, Tester DJ, et al. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome Circulation 2007;116:134-142. Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin–B mutation causes type 4 long–QT cardiac arrhythmia and sudden cardiac death. Nature 2003; 421: 634–9. Mohler PJ, Splawski I, Napolitano C, et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci USA 2004;101:9137–9142. Moss AJ, Liu JE, Gottlieb S, et al. Efficacy of permanent pacing in the management of high-risk patients with long QT syndrome. Circulation 1991;84:15241529 Moss AJ, Zareba W, Benhorin J, et al. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation. 1995;92:2929-2934. Moss AJ, Zareba W, Hall WJ, Schwartz PJ, Crampton RS, Benhorin J, Vincent GM, Locati EH, Priori SG, Napolitano C, Medina A, Zhang L, Robinson JL, Timothy K, Towbin JA, Andrews ML. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616–623. Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation 2000;101:616–23. Moss AJ, Zareba W, Kaufman ES, et al. Increased risk of arrhythmic events in long–QT syndrome with mutations in the pore region of the human ether–a–go–go–related gene potassium channel. Circulation 2002; 105: 794–799. Moss AJ, Shimizu W, Wilde AA, et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene Circulation 2007;115:2481-2489. Moss AJ, Zareba W, Schwarz KQ, et al. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J Cardiovasc Electrophysiol 2008;19:1289-1293. Peterson BZ, DeMaria CD, et al. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. Plaster NM, Tawil R, Tristani–Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell 2001; 105: 511–19. Potet F, Chagot B, Anghelescu M, et al. Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J Biol Chem. 2009;284:8846–8854. Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long–QT syndrome: clinical impact. Circulation 1999; 99: 529–33. Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long–QT syndrome. N Engl J Med 2003;348:1866–74. Priori SG, Napolitano C, Schwartz PJ, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA 2004;292:1341–1344. Ruan Y, Liu N, Bloise R, Napolitano C, et al. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation. 2007;116:1137-44. Ruan Y, Denegri M, Liu N, et al. Trafficking defects and gating abnormalities of a novel SCN5A mutation question gene-specific therapy in long QT syndrome type 3. Circ Res 2010;106:1374–1383. Sanguinetti MC. HERG1 channelopathies. Pflugers Arch. 2010;460:265-276. Schwartz PJ, Moss AJ, Vincent GM, et al. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993; 88: 782–4. Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation 1995;92:3381–3386. Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype–phenotype correlation in the long–QT syndrome: gene–specific triggers for life–threatening arrhythmias. Circulation 2001;103: 89–95. Schwartz PJ, Priori SG, Cerrone M, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation. 2004;109:1826–1833. Schwartz PJ, Spazzolini C, Crotti L, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation. 2006;113:783-790. Schwartz PJ, Stramba-Badiale M, Crotti L, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–1767. Schwartz PJ, Crotti L, Insolia R. Long-QT Syndrome. From Genetics to Management. Circ: Arrhythmia Electrophysiol 2012;5:868-877. Shamgar L, Ma L, Schmitt N, et al. Calmodulin is essential for cardiac IKS channel gating and assembly: impaired function in long-QT mutations. Circ Res. 2006;98:1055–1063. Shimizu W, Tanabe Y, Aiba T, et al.Differential effects of beta-blockade on dispersion of repolarization in the absence and presence of sympathetic stimulation between the LQT1 and LQT2 forms of congenital long QT syndrome. J Am Coll Cardiol. 2002;39:1984–91. Shimizu W, Noda T, Takaki H, et al. Diagnostic value of epinephrine test for genotyping LQT1, LQT2, and LQT3 forms of congenital long QT syndrome. Heart Rhythm 2004; 1: 276–83. Shimuzu W., Horie M., Ohno S. Mutation site-specific differences in arrhythmic risk and sensitivity to sympathetic stimulation in the LQT1 form of congenital long QT syndrome: multicenter study in Japan. J Am Coll Cardiol. 2004;44:117–125. Shimizu W, Moss AJ, Wilde AA, et al. Genotype-Phenotype Aspects of Type 2 Long QT Syndrome. J Am Coll Cardiol 2009;54:2052 -2062. Splawski I; Tristani-Firouzi M, Lehmann MH, et al. Mutations in the hmink gene cause long QT syndrome and suppress iks function. Nat. Genet. 1997;17:338–340. Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004; 119: 19–31. Shushi L, Kerem B, Goldmit M, et al. Clinical, genetic, and electrophysiologic characteristics of a new PAS-domain HERG mutation (M124R) causing long QT syndrome. Ann Noninvasive Electrocardiol. 2005;10:334–341. Sy RW, van der Werf C, Chattha IS, et al. Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation. 2011;124: 2187–2. Takenaka K, Ai T, Shimizu W, et al. Exercise stress test amplifies genotype–phenotype correlation in the LQT1 and LQT2 forms of the long–QT syndrome. Circulation 2003; 107: 838–44. Terrenoire C, Clancy CE, Cormier JW, et al. Autonomic control of cardiac action potentials: role of potassium channel kinetics in response to sympathetic stimulation. Circ Res. 2005;96:e25–e34. Tester DJ, Will ML, Haglund CM, et al. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2005;2:507–517 Tester DJ, Will ML, Haglund CM, Ackerman MJ. Effect of clinical phenotype on yield of long QT syndrome genetic testing. Am Coll Cardiol. 2006;47:764–768. Ueda K, Valdivia C, Medeiros-Domingo A, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci US A. 2008;105:9355-9360. Vatta M, Ackerman MJ, Ye B, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome Circulation 2006;114:2104-2112. Vincent GM, Schwartz PJ, Denjoy I, et al. High efficacy of beta-blockers in long-QT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures.” Circulation 2009;119:215–21. Viskin S, Fish R, Zeltser D, et al. Arrhythmias in the congenital long QT syndrome: how often is torsade de pointes pause dependent? Heart 2000; 83: 661–6. Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. Westenskow P, Splawski I, Timothy KW, et al. Compound mutations: a common cause of severe long–QT syndrome. Circulation 2004; 109: 1834–1841. Wilde A, Moss A, Kaufman E, et al. Clinical Aspects of Type 3 Long-QT Syndrome. An International Multicenter Study. Circulation. 2016;134:872–882. Yan GX, Wu Y, Liu T, Wang J, et al. Phase 2 early afterdepolarization as a trigger of polymorphic ventricular tachycardia in acquired long-QT syndrome: direct evidence from intracellular recordings in the intact left ventricular wall. Circulation 2001;103:2851-2856. Yang Y, Yang Y, Liang B, et al. Identification of a Kir3.4 mutation in congenital Long QT syndrome. Am J Hum Genet 2010;86:872–880. Yarbrough TL, Lu T, Lee HC, Shibata EF.Localization of cardiac sodium channels in caveolin-rich membrane domains: regulation of sodium current amplitude. Circ Res. 2002;90:443–449 Yoon G, Oberoi S, Tristani–Firouzi M, et al. Andersen–Tawil syndrome: Prospective cohort analysis and expansion of the phenotype. Am J Med Genet A 2006; 140: 312–21. Zareba W, Moss AJ, Schwartz PJ, et al. Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998;339:960–965. Zhang L, Timothy KW, Vincent GM, et al. Spectrum of ST–T–wave patterns and repolarization parameters in congenital long–QT syndrome: ECG findings identify genotypes. Circulation 2000; 102: 2849–55. Zhao JT, Hill AP, Varghese A, et al. Not All hERG pore domain mutations have a severe phenotype: G584S has an inactivation gating defect with mild phenotype compared to G572S, which has a dominant negative trafficking defect and a severe phenotype. J Cardiovasc Electrophysiol. 2009;20:923–930. Zhou Z, Gong Q, Epstein ML, et al. HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. J Biol Chem. 1998;273:21061–21066. Zhu W, Mazzanti A, Voelker TL, et al. Predicting Patient Response to the Antiarrhythmic Mexiletine Based on Genetic Variation. Circ Res. 2019 Feb 15;124(4):539-552. Zimmer T, Surber R. SCN5A channelopathies-an update on mutations and mechanisms. Prog Biophys Mol Biol 2008;98:120-136. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

|
|
| Aviso legal Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional |
