Canales de Sodio
|
Estructura La fase de rápida despolarización (fase 0) de los potenciales de acción de las células musculares auriculares y ventriculares y del sistema His-Purkinje es debida a la entrada de Na+ a través de canales activados por cambios de voltaje que generan la corriente INa. Esta corriente determina la excitabilidad y la velocidad de conducción de los PA a través de las aurículas y los ventrículos (George 2005). Además, los canales de Na+ dependientes de voltaje son el receptor de los anestésicos locales, algunos antiepilépticos y los fármacos antiarrítmicos del grupo I. Los canales de Na+ voltaje-dependientes están compuestos por una subunidad α (Nav1.5, 260 kDa) codificada por el gen SCN5A y una o más subunidades β (Navβ1.-4; 33-36 kDa) codificadas por los genes SCN1B-SCN4B (Goldin, 2002). La mayoría de los canales de Na+ cardíacos son resistentes a la tetrodotoxina (TTX) (KD, 2-6 6 μM), aunque también se ha demostrado la expresión en el miocardio de otras subunidades α neuronales (Nav1.1, Nav1.3 y Nav1.6) sensibles a tetrodotoxina (KD = 1-10 nM) cuya función aún no está del todo clara (Nerbonne y Kass, 2005), aunque podrían jugar un papel en la actividad marcapaso del nodo SA (Lei et al., 2007). La localización subcelular de la subunidad α a nivel de los discos intercalares confirman su importante papel en la regulación de la conducción cardiaca (Kucera et al., 2002). Es un complejo de heteromultimérico proteico integral que consta de cuatro dominios homólogos (D1-D4), cada uno de los cuales contiene seis segmentos α-hélice transmembrana (S1-S6, de 19 a 27 aminoácidos). Los extremos C-y N-terminal y los lazos de unión de los dominios entre sí son intracitoplasmáticos. Los segmentos S5-S6 y el lazo P de cada dominio forman el poro del canal que penetra en el interior de la membrana y contiene el filtro de selectividad del mismo (Yamagishi et al, 2001; George, 2005; Yu y Catterall, 2003). La porción citoplásmica del poro está formada por la combinación de los segmentos S5 y S6 de los cuatro dominios. La mayor parte de los residuos que forman los segmentos S5 y S6 son hidrófobos, mientras que en los S1-S3 hay pocos residuos cargados (20 cargas negativas). Sin embargo, en el segmento S4 uno de cada tres residuos está cargado (arginina o lisina: 4 en DIS4, 5 en DIIS4 y DIIIS4 y 8 en el DIVS4), razón por la que el segmento S4, que funciona como un sensor de voltaje (Stühmer et al, 1989; George, 2005; Yu y Catterall, 2003). Durante la despolarización alrededor de 12 de las 22 cargas que forman parte del sensor de voltaje, se desplazan siguiendo un movimiento en espiral a través del campo eléctrico transmembrana, iniciando así un cambio conformacional que facilita la apertura del canal de Na+ (Stuhmer et al., 1989). Como consecuencia, 3 cargas positivas del segmento S4 del dominio DIV, que previamente estaban embebidas en la membrana, quedan expuestas en la superficie externa y otras 2 cargas quedan expuestas en la superficie interna (Yang y Horn, 1995; Catterall, 2000). El lazo citoplásmico entre los dominios DIII y DIV contiene una secuencia de isoleucina-fenilalanina-metionina (IFM), que actúa como una partícula de inactivación que ocluye el poro del canal, dando lugar a la inactivación del mismo tras su apertura (Stuhmer et al., 1989 Patton et al., 1992, West et al., 1992). La subunidad α contiene también el receptor para anestésicos locales, antiarrítmicos y anticonvulsivos, formado por residuos localizados en el segmento S6 de los dominios DI, DII y DIV.
Aunque la subunidad α formadora del poro es suficiente para la expresión funcional del canal de Na+, la cinética y la dependencia de voltaje de la activación de canales es modificada por las subunidades β y que también están involucradas en la localización del canal y por la interacción con moléculas de adhesión celular, matriz extracelular y citoesqueleto intracelular (Caterall et al., 2019). Existen cuatro subuniddaes β diferentes subunidades (33-36 kDa) codificadas por los genes SCN1B, SCN2B, SCN3B y SCN4B, respectivamente Se trata de glicoproteínas transmembrana de tipo 1 con un único dominio transmembrana, un extremo N-extracelular y un extremo C-citoplasmático (Brackenbury e Isom, 2011). El extremo extracelular tiene una estructura homóloga a la superfamilia de inmunoglobulinas Ig), lo que sugiere que las subunidades pueden actuar como moléculas de adhesión celular para fijar los canales al sarcolema y facilitarían la interacción de los canales de Na+ con gran una variedad de moléculas de señalización (Makita et al., 1996; Isom, 2001, Brackenbury e Isom, 2011). Las subunidades β aumentan el tráfico del canal hacia el sarcolema, aumentan la amplitud de INa, modulan sus propiedades de activación de puerta (dependencia de voltaje de la (in)activación) y desempeñan un papel crucial en la interacción de las proteínas Nav1.5 con otras moléculas de la matriz extracelular, del citoesqueleto y con otros componentes de las uniones intercelulares cardíacas (p. ej., cadherinas, conexinas). También pueden contribuir a la localización preferencial de las proteínas Nav1.5 en los discos intercalados. Otras proteínas con las que interactúan Muchas proteínas interactúan de forma transitoria con los canales de Na+ en el contexto de la regulación de los canales, incluidas varias proteínas quinasas y proteínas G, mientras que otras interactúan de forma más permanente, incluidas las moléculas de adhesión celular y las proteínas del citoesqueleto (Caterall et al., 2019). La calmodulina se une a un sitio conservado en el dominio C-terminal proximal del canal de Na+ y participa en la inactivación del mismo (Gabelli et al., 2014). Los factores homólogos (FHF) del factor de crecimiento de fibroblastos también pueden formar un complejo estable con el extremo C-terminal de los canales de Na+ y las mutaciones localizadas en la zona de enlace entre Nav1.5 y FHF (p.H1849R) producen una función de ganancia de función y aumentan la INa al alterar la inactivación en estado estable y disminuir la velocidad de inactivación de Nav1.5 y los miocitos que expresan Nav1.5 p.H1849R presentaban una duración del potencial de acción prolongada y posdepolarizaciones arritmogénicas (Musa et al., 2015). Estados conformacionales del canal de sodio
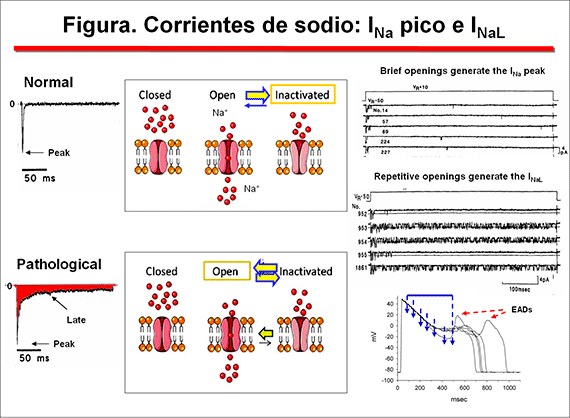
Durante el potencial de acción (PA) cardiaco, los canales de Na+ adoptan tres estados conformacionales: uno conductor (abierto-O) y dos no conductores (cerrado-C, inactivo-I)(Caterall, 2000; Balser, 2001) (figura 2). La transición entre es tos estados es un proceso voltaje- y tiempo-dependiente que se conoce como gating del canal. Durante la diástole el canal se encuentra en un estado de C no-conductor o de reposo y la probabilidad de que se abra es extremadamente baja. Durante la despolarización del potencial de membrana (fase 0 del PA) se produce un rápido aumento de la permeabilidad al Na+ debido a la apertura (activación) de los canales desde el estado C (C → O), permitiendo la entrada de Na+ a su través durante 1-2 ms, lo que genera la corriente rápida de entrada de Na+ (INa). Modelos de cómo funciona el sensor de voltaje predicen que el segmento S4 se desliza 6-8 Å hacia afuera a través de un estrecho surco formado por los segmentos S1, S2 y S3, gira ∼ 30° y se inclina hacia los lados en un punto de giro formado por una región hidrófoba altamente conservada localizada cerca de la mitad del sensor de voltaje. El segmento S4 tiene una conformación helicoidal 3(10) en el estrecho poro interno, lo que permite el movimiento lineal de las cargas de la puerta a través de la mitad interna de la membrana. Los cambios conformacionales de la mitad intracelular de S4 durante la activación se acoplan al movimiento lateral del lazo S4-S5, lo que podría inducir el movimiento de los segmentos S5 y S6 y abrir la puerta intracelular del poro (Yarov-Yarovoy et al., 2012).
La INaL generada a través de canales abiertos durante la fase de meseta del PA (es decir, fase 2) puede incrementarse notablemente en condiciones patológicas (p.ej., isquemia, insuficiencia cardíaca, SQTL3), lo que lleva a una prolongación de la DPA ventricular (intervalo QT del ECG) (Zaza et al., 2008). Las mutaciones en el enlazador entre los dominios DIII y DIV en SCN5A, vinculados al SQTL3, interrumpen la inactivación del canal Na+ (Bennett et al., 1995). Estas mutaciones de "ganancia de función" aumentan la amplitud de INaL, prolongan la DPA y pueden permitir el desarrollo de actividad desencadenada (triggered focal activity) disparada por la aparición de postpotenciales tempranos.
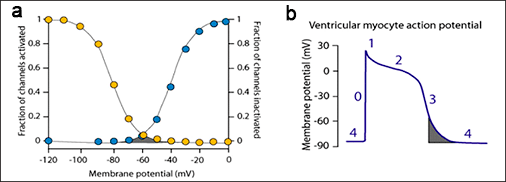
Figura. Corriente ventana de sodio. a) La corriente de la ventana (área gris) está determinada por el cruce de las curvas de activación e inactivación que gobiernan la apertura del canal de sodio. b) Este entrecruzamiento tiene lugar durante la última fase 3 de repolarización.
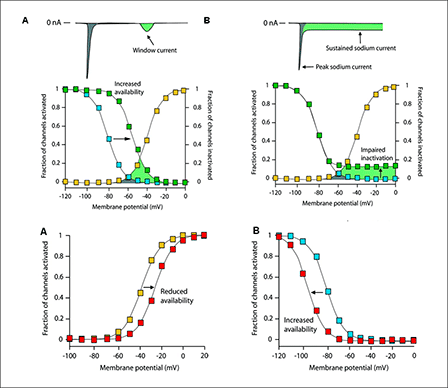
Figura. Mutaciones en el gen SCN5A que codifica la subunidad α del canal de Na+ modician el gating del canal.
Varias síndromes arritmogénicos primarios que se transmiten de forma autosómica dominante y se asocian a mutaciones puntuales en los genes que codifican la subunidad 1 de los canales Nav1.5 cardíacos [p.ej., SQTL (SQTL 3, 9, 10, 12), SBr1, SRT, FA idiopática, defectos progresivos de la velocidad de conducción intracardiaca, parada auricular, síndrome del seno enfermo congénita, síndrome de muerte súbita del lactante y algunos tipos de cardiomiopatía dilatada arritmogénica]( George, 2005; Clancy y Kass, 2005).
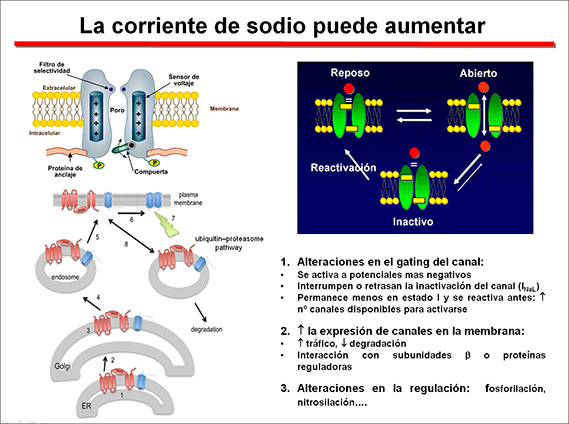
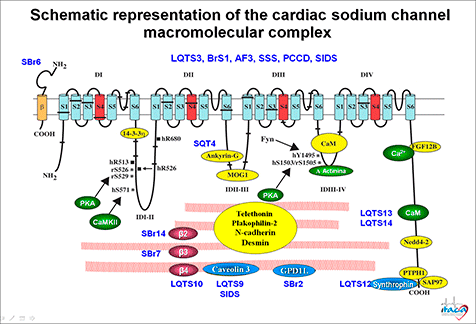
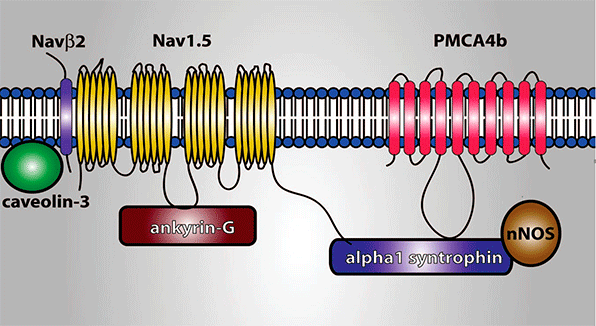
En la Figura se muestra el conjunto de proteínas que hoy en día se considera que constituyen el “canalosoma” de los canales de Na cardiacos humanos. Se puede observar que, además de las subunidades α (Nav1.5) y β (Navβ 2), el canal está unido a otras proteínas que las unen al citoesqueleto [tales como actina, caveolina-3, anquirina G, placofilina, α-sintrofina o diversas cadherinas], a proteínas que participan en el tráfico de las subunidades desde el retículo endoplásmico (ER) al sarcolema (p.ej., la glicerol-3-fosfato deshidrogenasa 1 o GPD1-L). A su vez, el canal puede ser modulado por múltiples vías de señalización, incluidas las enzimas involucradas en su glicosilación y fosforilación [Ca2+/calmodulina proteína cinasa II (CaMKII), calmodulina, proteína cinasa A (PKA) o las proteínas 13-3-3]. Estas moléculas reguladoras y de señalización juegan un importante papel en la regulación del tráfico y de la expresión de los canales en la membrana, así como en el funcionamiento del canal (Xiao et al, 1999; Ratcliffe et al, 2001; Mohler et al., 2002).
Figura. Esquema en el que se representan las diversas proteínas del canalosoma del canal de Na+ cardiaco humano. Amin A, Asghari-Roodsari A, Tan HL. Cardiac sodium channelpathies. Pfluggers Arch-Eur J Physiol 2010;460:223-237. Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70:567-90. Armstrong CM. Sodium channel and gating currents. Physiol Rev 1981;61:644-682. Attwell D, Cohen I, Eisner D, Ohba M, and Ojeda C. The steady state TTX-sensitive (“window”) sodium current in cardiac Purkinje fibres. Pflügers Arch 1979;379:137-142. Balser JR. The cardiac sodium channel: gating function and molecular pharmacology. J Mol Cell Cardiol 2001;33:599-613. Bennett PB, Yazawa K, Makita N, George AL Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature 1995;76:683–685. Brackenbury WJ, Isom LL. Na Channel β Subunits: Overachievers of the Ion Channel Family. Front Pharmacol. 2011;2:53. Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 2000;26:13–25. Catterall WA, Swanson TM. Structural basis for pharmacology of voltage-gated sodium and calcium channels. Mol Pharmacol 2015;88:151-150. Catterall WA, Goldin AL, Waxman SG. Voltage-gated sodium channels. Accessed on 22/07/2019. IUPHAR/BPS Guide to PHARMACOLOGY, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=82. 2019. Chen CJ, Juang JJ, Lin LY, et al; (SADS-TW BrS Registry) . Gender difference in clinical and genetic characteristics of Brugada syndrome: SADS-TW BrS registry. QJM. 2019;112:343-350. Clancy CE, Kass RS. Inherited and acquired vulnerability to ventricular arrhythmias: cardiac Na+ and K+ channels. Physiol Rev. 2005;85:33-47. Eaholtz G, Scheuer T, Catterall WA. Restoration of inactivation and block of open sodium channels by an inactivation gate peptide. Neuron. 1994;12:1041-8. Fahmi AI, Patel M, Stevens EB, et al. The sodium channel beta-subunit SCN3B modulates the kinetics of SCN5A and is expressed heterogeneously in sheep heart. J Physiol 2001;537:693-700. Gabelli SB, Boto A, Kuhns VH, et al. Regulation of the NaV1.5 cytoplasmic domain by calmodulin. Nat Commun 2014;5:5126. George AL. Inherited disorders of voltage-gated sodium channels. J Clin Invest 2005;115:1990-1999. Goldin AL. Evolution of voltage-gated Na(+) channels. J Exp Biol. 2002;205:575-84. Grant AO, Carboni MP,Neplioueva V, et al. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J Clin Invest 2002;110:1201–1209. Heinemann SH, Terlau H, Stühmer W, et al., Calcium channel characteristics conferred on the sodium channel by single mutations. Nature 1992;356:441–443. Isom LL. Sodium channel beta subunits: anything but auxiliary Neuroscientist. 2001;7:42-54. Kellenberger S, Scheuer T, Catterall WA. Movement of the Na+ channel inactivation gate during inactivation. J Biol Chem. 1996;271:30971-9. Kucera JP, Rohr S, Rudy Y. Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ Res. 2002;91:1176-82. Lei M, Zhang H, Grace AA, et al. SCN5A and sinoatrial node pacemaker function. Cardiovasc Res 2007;74:356-65. Makielski JC, Limberis JT, Chang SY, et al. Coexpression of β1 with cardiac sodium channel alpha subunits in oocytes decreases lidocaine block. Mol Pharmacol 1996;49:30-39. Makita N, Bennett PB, George AL Jr. Molecular determinants of β1 subunit-induced gating modulation in voltage-dependent Na+ channels. J Neurosci 1996;16:7117-7127. McPhee JC, Ragsdale DS, Scheuer T, et al. A critical role for transmembrane segment IVS6 of the sodium channel alpha subunit in fast inactivation. J Biol Chem 1995;270:12025-12034. McPhee JC, Ragsdale DS, Scheuer T, et al. A critical role for the S4–S5 intracellular loop in domain IV of the sodium channel alpha-subunit in fast inactivation. J Biol Chem 1995;273:1121-1129. McPhee JC, Ragsdale DS, Scheuer T, et al. A critical role for the S4-S5 intracellular loop in domain IV of the sodium channel alpha-subunit in fast inactivation J Biol Chem 1998;273:1121-9. Mohler PJ, Gramolini AO, and Bennett V. Ankyrins. J Cell Sci 2002;115:1565-1566. Motoike HK, Liu H, Glaaser IW, et al. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J Gen Physiol 2004;123:155-165. Musa H, Kline CF, Sturm AC, et al. SCN5A variant that blocks fibroblast growth factor homologous factor regulation causes human arrhythmia. Proc Natl Acad Sci U S A. 2015;112:12528-33. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolariation. Physiol Rev 2005;85:1205-1253. Noda M, Suzuki H, Numa S, et al. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett 1989;259:213–216.Patton DE, West JW, Catterall WA, et al. Amino acid residues required for fast Na_-channel inactivation: charge neutralizations and deletions in the III-IV linker. Proc Natl Acad Sci USA 1992;89:10905–10909. Ratcliffe CF, Westenbroek RE, Curtis R, et al. Sodium channel beta1 and beta3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol 2001;154:427-434. Rohl CA, Boeckman FA, Baker C, et al. Solution structure of the sodium channel inactivation gate. Biochemistry. 1999;38:855-61. Stühmer W, Conti F, Suzuki H, et al. Structural parts involved in activation and inactivation of the sodium channel. Nature 1989;339:597-603. Terlau H, Heinemann SH, Stühmer W, et al. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett 1991;293:93–96. Ueda K, Valdivia C, Medeiros-Domingo A, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355-60. Ulbricht W. Sodium Channel Inactivation: Molecular determinants and modulation. Physiol Rev 2005;85:1271-1301. Vassilev P, Scheuer T, Catterall WA. Inhibition of inactivation of single sodium channels by a site-directed antibody. Proc Natl Acad Sci U S A. 1989;86:8147-51. West JW, Patton DE, Scheuer T, et al. A cluster of hydrophobic amino acid residues required for fast Na_-channel inactivation. Proc Natl Acad Sci USA 1992;89:10910–10914. Wilde AA, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res. 2011;108:884-97. Xiao ZC, Ragsdale DS, Malhotra JD, et al. Tenascin-R is a functional modulator of sodium channel beta subunits. J Biol Chem 1999;274:26511-26517. Yamagishi T, Li RA, Hsu K, Marbán E, et al. Molecular architecture of the voltage-dependent Na channel: functional evidence for alpha helices in the pore. J Gen Physiol 2001;118:171-82. Yang N, Horn R. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 1995;15:213-8. Yarov-Yarovoy V, DeCaen PG, Westenbroek RE, et al. Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci USA 2012;109:E93–E102. Yu FH, Catterall WA. Overview of the voltage-gated sodium channel family.Genome Biol. 2003;4:207. Zaza A, Belardinelli L, Shryock JC. Pathophysiology and pharmacology of the cardiac "late sodium current". Pharmacol Ther 2008;119:326-339.
|
|
|
| Aviso legal Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional |
