Canales voltaje-dependientes de Ca2+ Tipo-L
|
Estructura del canal
En cardiomiocitos en reposo, la concentración intracelular de Ca2+ ([Ca2+]i) es 20.000 veces menor que su concentración en el medio extracelular (<0.1 μM vs 1-2 mM) y el interior celular es electronegativo (-50 a -90 mV). Es decir, que existe un gradiente electroquímico que favorece la entrada de iones Ca2+ en la célula. Durante la activación celular, la [Ca2+]i aumenta hasta 0.3-1 μM como consecuencia de la entrada de Ca2+ desde el medio extracelular a través de la membrana, bien a través de canales voltaje dependientes tipo-L (y, en mucho menor grado, del intercambiador Na+-Ca2+) y/o de la liberación de Ca2+ desde sus depósitos intracelulares, principalmente el retículo sarcoplásmico reticulum (Ca2+ induced Ca2+ release). Los canales de Ca2+ voltaje-dependientes tipo-L son heterotetrámeros compuestos por 4 subunidades: una subunidad α1C que forma el poro del canal (1C o 1D, 242 kDa) y distintas subunidades auxiliares, incluídas el dímero α2/δ (175 kDa) unidas por un puente disulfuro y las subunidades β1-3 (55-60 kDa) intracelulares (Figura) (Nerbonne y Kass, 2005; Bodi et al., 2005). Estas subunidades se condifican, respectivamente, por los genes CACNA1C o CACNA1D, CACNA2D1 o CACNA2D3 y CACNB1-3 (Nerbonne y Kass, 2005; Bodi et al., 2005). Las subunidades β están estrechamente asociadas en la cara citoplásmica de la subunidad α1 (a través del lazo que une los dominios I-II), mientras que las subunidades α2δ están ancladas a la membrana plasmática e interactúan con dominios extracelulares de la sububidad α1. (Cav1.2). Está formada por 2179 aminoácidos que se organizan en 4 dominios homólogos (DI-DIV), cada uno de los cuales está compuesto por 6 segmentos transmembrana (S1-S6). Esta subunidad contiene el poro iónico, que está formado por el lazo P que une S5 y S6 en cada dominio y penetra en la membrana, el filtro de selectividad, los mecanismos que regulan la cinética de apertura y cierre del canal y los puntos de unión para la subunidad α y para los bloqueantes de los canales de Ca2+ (IIIS5, III6 andIVS6). Al igual que el canal de Na+ voltaje-dependiente, se cree que la α-hélice del S4, que contiene varios residuos con cargas positivas, forma el sensor de voltaje, que se mueve en el campo transmembrana iniciando unos cambios conformacionales que modulan la condutancia del canal (Bezanilla, 2002, Caterall, 2000). El análisis estructural ha permitido ientificar una serie de cinco sitios de unión para Ca2+, que van desde la entrada al vestíbulo exterior a través del filtro de selectividad hasta la cavidad central (Tang et al., 2014). Este hallazgo es cosnistente con un mecanismo knock-off, en el cual la unión de un ion Ca2+ bloquea el poro y evita la entrada de cationes monovalentes, mientras que la unión de un segundo Ca2+ a un segundo sitio de unión de Ca2+ induce la repulsión electrostática que empuja el primer Ca2+ unido a través del filtro de selectividad hacia el citosol. Este mecanismo explica cómo el canal de Ca2+ es altamente selectivo con respecto a otros cationes monovalentes y presenta una alta conductancia para el Ca2+, aunque los diámetros atómicos de Na+ y Ca2+ son casi idénticos (Caterall y Swanson, 2015).
Se han identificado 4 subunidades β (β1-4, 55 kDa) codificadas por los genes CACNB1, CACNB2, CACNB3, y CACNB4, respectivamente. En general, la coexpresión de las subunidades β modula las propiedades biofísicas de las subunidades α1, produciendo un desplazamiento hacia la izquierda de la relación corriente-voltaje y regulan la expresión del canal en la superficie de la membrana celular.
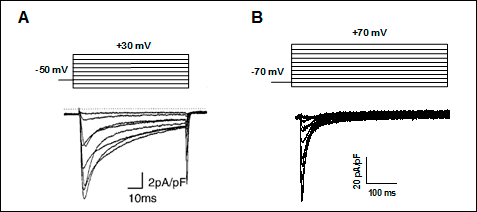
Figura 9. (A) ICa,L registrada en miocitos auriculares humanos tras la aplicación del protocolo que se muestra en la parte superior. Tomada de Van Wagoner et al., 1999. (B) Canales Cav1.2, β2 y α2/δ1 (tomado de Delpón y Tamargo). Nótese la diferencia de escala temporal entre los trazos de corriente de ambos paneles.
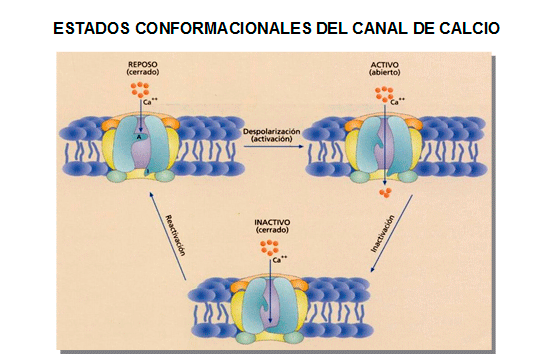
Los canales tipo-L se activan-abren cuando se despolariza el potencial de membrana a valores positivos a -40 mV y la corriente alcanza su máxima amplitud entre 0 y +10 mV. Los canales se activan-abren muy rápidamente durante la despolarización de la membrana y persisten abiertos durante toda la fase 2 del PA cardíaco, generando una corriente de entrada de Ca2+ o ICaL. La activación y la inactivación son relativamente lentas, alcanzándose el pico máximo al cabo de 1-7 ms y se inactiva con una inact > 500 ms. La superposición de las curvas de activación e inactivación producen una "ventana de ventana" similar a la descrita para los canales de Na+. Cuando el PA cardíaca se prolonga de forma excesiva (prolongación del intervalo QT) en potencial de membrana entra en un rango de potenciales (comprendido entre -15 y -40 mV) en el que la ICaL que ha sido inactivada en gran parte durante la fase de meseta del PA puede ser parcialmente reactivada según la [Ca2+]i disminuye. Esta reactivación puede causar una despolarización neta del potencial de membrana e inducir pospotenciales tempranos y actividad desencadenada (triggered focal activity).
La inactivación del canal de Ca2+ es un proceso regulado por dos mecanismos: uno voltaje-dependiente y otro Ca2+-dependiente (Kass y Sanguinetti, 1984; Lee et al., 1985). La activación de los canales Cav1.2 utilizando Ba2+ como el ion que atraviesa los canales de Ca2+ permite registrar corrientes de entrada de Ba2+ que se activan rápidamente e inactivan lentamente a través de un mecanismo voltaje-dependiente exclusivamente. Por el contrario, cuando el ión que se conduce a través de los caanles es el Ca2 las corrientes de Ca2+ generadas se inactivan rápidamente a través de una inactivación Ca2+/calmodulina-dependiente. La calmodulina se une al extremo C-terminal de la subunidad 1C en reposo, cerca del motivo IQ y que actúa como el sensor Ca2+. Durante la despolarización, el Ca2+ entra en las células cardiacas y se une a los sitios de unión de alta afinidad en el lóbulo C-terminal de la calmodulina, que reduce la inhibición mediada por la mano EF del lazo de unión entre los dominios I-II (que representa la partícula bloqueante del canal), permitiendo su interacción con el poro y la inactivación del canal (Kim et al., 2004). Por lo tanto, la calmodulina está implicada en la inactivación Ca2+-dependiente, un mecanismo regulador fisiológico que evita una entrada excesiva de Ca2+ en los cardiomiocitos que conduciría a la necrosis cardiaca (Qin et al., 1999, Zühlke et al., 1999).
La densidad de canales de Ca2+ tipo L aumenta con la edad y en diversas situaciones patológicas (hipertensión arterial, cardiomiopatías) y tras la ingesta de sal. Los fármacos que aumentan los niveles de AMPc (agonistas β-adrenérgicos, inhibidores de la fosfodiesterasa) activan la vía AMPc-PKA, que fosforila la subunidad 1C y aumentan la probabilidad de apertura de los canales tipo-L cardíacos y la ICaL. La activación de la PKC por ésteres de forbol o angiotensina II también aumenta la ICaL. Por el contrario, los fármacos que aumentan los niveles de GMPc (dadores de NO, acetilcolina) estimulan la PKG, que bloquea la ICa. Esta relación antagónica entre AMPc y GMPc permite al sistema nervioso vegetativo controlar la contractilidad y la frecuencia cardíacas. Los bloqueantes de los canales de Ca2+ inhiben de forma selectiva la ICaL. La acidosis, la isquemia cardíaca y la fibrilación auricular disminuyen la ICaL. Las mutaciones en el gen CACNA1C que codifica Cav1.2 están asociadas al síndrome de QT largo 8 (síndrome de Tymothy), síndrome de Brugada 3, síndrome de QT corto 4 y síndrome de repolarización temprana 2; mutaciones en el gen CACNA2D1 que codifica la subunidad 2 en el síndrome de QT corto 6 y en el síndrome de repolarización temprana 4 y las mutaciones en el gen CACNB2B que codifica la subunidad b2 en el síndrome de Brugada 4, el síndrome de QT corto 5 y el síndrome de repolarización temprana 3. Bangalore R, Mehrke G, Gingrich K, et al. Influence of L-type Ca channel alpha 2/delta-subunit on ionic and gating current in transiently transfected HEK 293 cells. Am J Physiol Heart Circ Physiol 1996;270:H1521–H1528. Bichet D, Cornet V, Geib S, et al. The I-II loop of the Ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron. 2000;25:177-190. Bodi I, Mikala G, Koch SE, et al. The L-type calcium channel in the heart: the beat goes on. J Clin Invest. 2005;115:3306-3317. Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000;16:521–555. Catterall WA, Swanson TM. Structural Basis for Pharmacology of Voltage-Gated Sodium and Calcium Channels. Mol Pharmacol 2015;88:141-145. Christel C, Lee A. Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim Biophys Acta. 2012; 1820:1243–1252. De Waard M, Pragnell M, Campbell KP. Ca2+ channel regulation by a conserved beta subunit domain. Neuron. 1994;13:495-503. Gurnett CA, De Waard M, Campbell KP. Dual function of the voltage-dependent Ca2+ channel alpha 2 delta subunit in current stimulation and subunit interaction. Neuron 1996;16:431–440. Hockerman GH, Peterson BZ, Johnson BD, et al. Molecular determinants of drug binding and action on L-type calcium channels. Annu Rev Pharmacol Toxicol. 1997;37:361–396. Hofmann F, Biel M, Flockerzi V. Molecular basis for Ca2+ channel diversity. Annu Rev Neurosci. 1994;17:399-418. Hulme JT, Lin TW, Westenbroek RE, et al. Beta-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci U S A. 2003;100:13093-8. Hulme JT, Yarov-Yarovoy V, Lin TW, et al. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J Physiol. 2006;576:87-102. Kass RS, Sanguinetti MC. Inactivation of calcium channel current in the calf cardiac Purkinje fiber. Evidence for voltage- and calcium-mediated mechanisms. J Gen Physiol 1984;84:705-726 Kim J, Ghosh S, Nunziato DA, et al. Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron 2004a;41:745-754. Lee KS, Marbán E, Tsien RW. Inactivation of calcium channels in mammalian heart cells: joint dependence on membrane potential and intracellular calcium. J Physiol (Lond.) 1985;364:395-411. Mikala G, Klockner U, Varadi M, et al. cAMP-dependent phosphorylation sites and macroscopic activity of recombinant cardiac L-type calcium channels. Mol Cell Biochem 1998;185:95-109. Mori Y, Mikala G, Varadi G, et al. Molecular pharmacology of voltage-dependent calcium channels. Jpn J Pharmacol. 1996;72:83-109. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev 2005;85:1205-125 Perez-Reyes E, Castellano A, Kim HS, et al. Cloning and expression of a cardiac/brain beta subunit of the L-type calcium channel. J Biol Chem 1992;267:1792-1797. Pragnell M, De Waard M, Mori Y, et al. Calcium channel betasubunit binds to a conserved motif in the I-II cytoplasmic linker of the alpha 1-subunit. Nature 1994;368:67-70. Singer D, Biel M, Lotan I, et al. The roles of the subunits in the function of the calcium channel. Science 1991;253:1553–1557. Tang L, Gamal El-Din TM, et al. Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature 2014;505:56-61. Van Petegem F, Clark KA, Chatelain FC, et al. Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature. 2004;429:671-5. Van Wagoner DR, Pond AL, Lamorgese M, et al. Atrial L-type Ca2+ currents and human atrial fibrillation. Circ Res. 1999;85:428-36. Varadi G, Lory P, Schultz D, et al. Acceleration of activation and inactivation by the beta subunit of the skeletal muscle calcium channel. Nature. 1991;352:159-62. Viard P, Butcher AJ, Halet G, et al. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat. Neurosci. 2004;7:939–946. Wei SK, Colecraft HM, DeMaria CD, et alT. Ca2+ channel modulation by recombinant auxiliary beta subunits expressed in young adult heart cells. Circ Res 2000;86:175-184. Yamaguchi H, Hara M, Strobeck M,et al. Multiple modulation pathways of calcium channel activity by a beta subunit. Direct evidence of beta subunit participation in membrane trafficking of the alpha1C subunit. J Biol Chem 1998;273:19348–19356. Zamponi GW, Striessnig J, Koschak A, et al. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol Rev 2015;67:821-70 Zuhlke RD, Pitt GS, Deisseroth K, et al. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature 1999;399:159-162.
|
|
|
| Aviso legal Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial-SinObraDerivada 4.0 Internacional |
